此是记录的按照前面做了以后
废话不多说,先调整周期性
全部选择
0(“System”)
老规矩,作图
|
|
全部选择碳链骨架
4(“Backbone”)
xmgrace作图以后,让人紧张的情绪膨胀
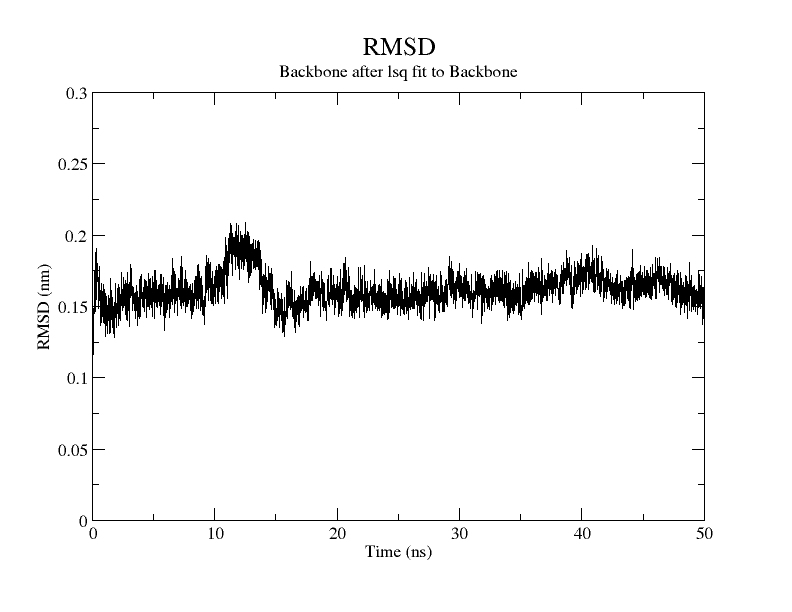
也不知道后面是不是真的是稳定了的说
2.rmsd
|
|
3.rmsf
|
|
如图:
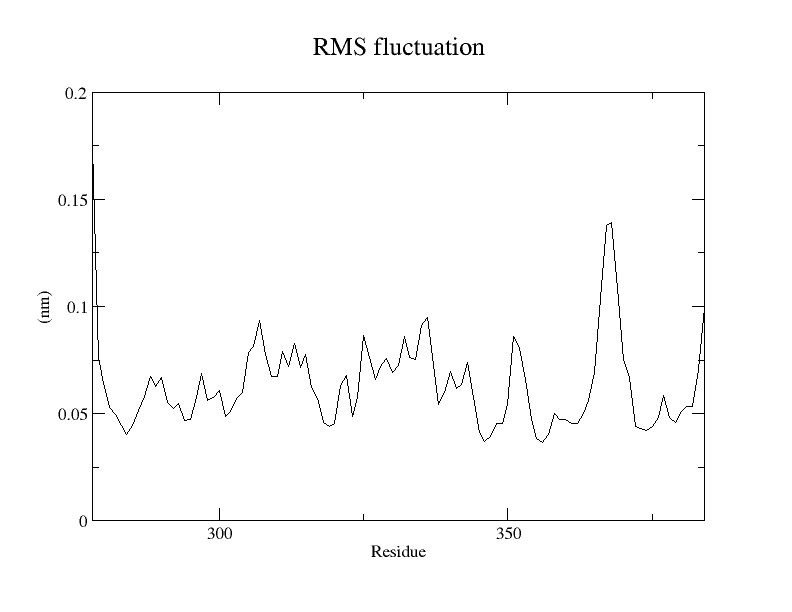
可以发现越是突变位点越发稳定
4.g_confrms比较结构差异
|
|
但是不知道为何生成的fit.pdb文件出来有一点问题,所以想尝试第二种方案
|
|
在pymol中打开md_0_1.pdb,
|
|
得到的结果图用cartoon作图结果如下:
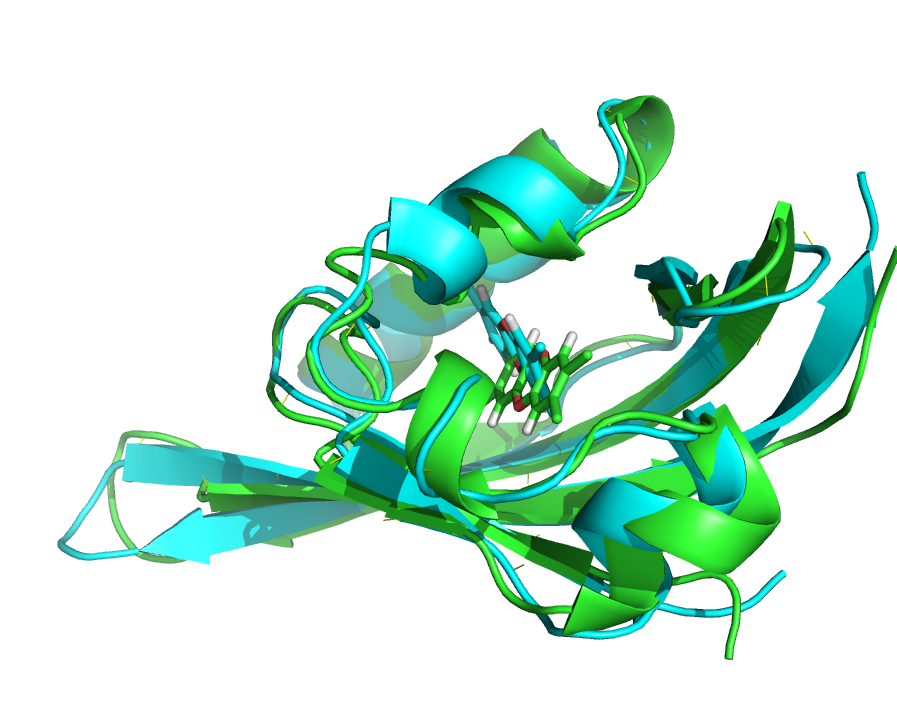
可以看出结构垭口和loop环的差别还是很大的,TCDD有一个翻转并且进一步深入口袋
5.豪猪图
采用Pymol画豪猪图,方法如前文
关键命令如下
|
|
结果如下:
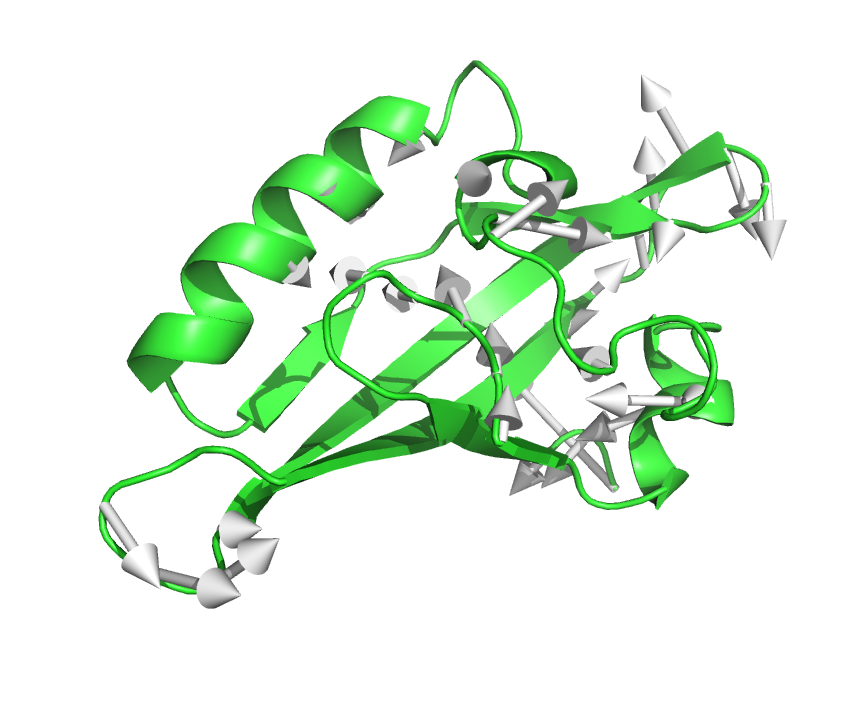
可以看出50ns AhR蛋白beta折叠和α螺旋并未进行剧烈的变动,仅仅为loop环结构使得接合口袋进一步的收缩,轻微的构象改变更好的说明同源建模过程中构象的准确性。
6.查看小分子的运动轨迹
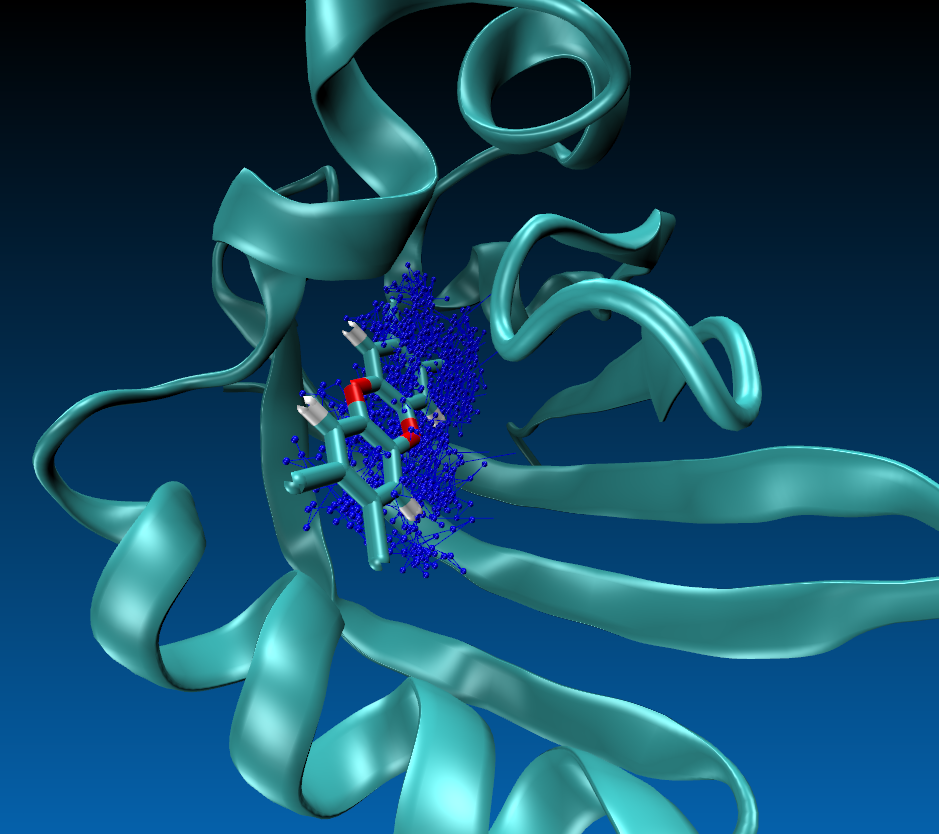
小圆点代表的为tcdd分子中每个原子1ns行走的位置,可以发现tcdd在接合口袋相当稳定,在一定波动范围波动,稍稍深入蛋白腔内部。
7.回旋半径的计算
蛋白质的回旋半径反应了蛋白质分子的体积和形状。同一体系的回旋半径越大,说明体系发生了膨胀。
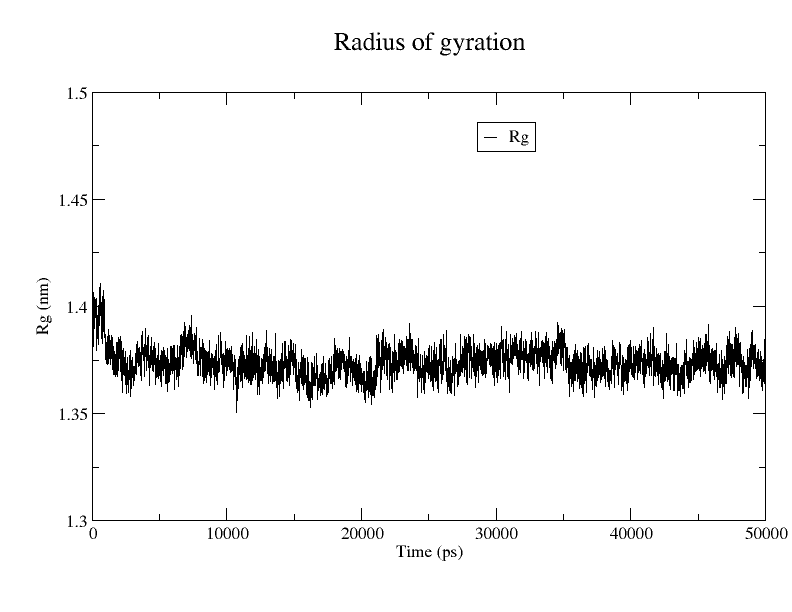
发现体系的回旋半径很稳定。
|
|
8.dssp二级结构
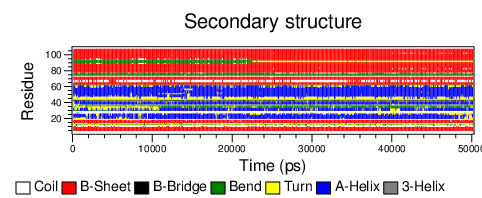
可以看出蛋白结构还是比较稳定,具体需要进一步研究
|
|
9.计算最大螺旋性质
g_helix_mpi -s md_0_1.tpr -n index.ndx -f mdwholeNOjump.xtc -dt 100
gmx helix计算各种类型螺旋的性质. 程序首先会检查多肽段, 找到最长的螺旋部分, 这由氢键和φ/ψ角度确定的. 再将其拟合成一个绕z轴的理想螺旋, 以原点居中. 然后计算以下性质:
螺旋半径(radius.xvg输出文件). 这仅仅是二维平面内所有C~α~原子的RMS偏差, 计算方法sqrt((sum_i (x\^2(i)+y\^2(i)))/N), 其中N为骨干原子数. 理想螺旋的半径为0.23 nm.
扭转(twist.xvg输出文件). 计算每个残基的平均螺旋角. 对α螺旋此值为100度, 对3-10螺旋值会更小, 5-螺旋的值更大.
每个残基的上升量(rise.xvg输出文件). 每个残基的螺旋上升量以C~α~原子z坐标的差值表示. 对于理想螺旋此值为0.15 nm
总螺旋长度(len-ahx.xvg输出文件). 以nm为单位的总螺旋长度. 其值简单由平均上升量(见上文)乘上螺旋残基数(见下文)计算.
螺旋偶极. 只计算骨干原子的(dip-ahx.xvg输出文件).
与理想螺旋的RMS偏差, 仅根据C~α~原子计算(rms-ahx.xvg输出文件).
平均C~α~-C~α~二面角(phi-ahx.xvg输出文件)
平均φ和ψ角度(phipsi.xvg输出文件).
根据Hirst和Brooks方法计算的222 nm处的椭圆度
10.矩阵分析(PCA)
|
|
待编辑
参考文献:水中的溶菌酶教程
参考文献:漏斗网蜘蛛毒素肽的溶剂化研究: Amber99SB-ILDN力场
参考文献:dssp二级结构
参考文献:gromacs中的do_dssp的使用

