该教程计划每周自己做一个
转载请联系,且保留链接
翻译自:Trieste tutorial: Analyzing trajectories using PLUMED
目标 主要是熟悉PLUMED语句,并用来写简单的CV(collective variable)进行分析已有的轨迹
流程
写一个简单的PLUMED输入文件,使用PLUMED driver来分析轨迹
使用GROUP关键字进行压缩和快速的构建复杂的原子组
打印PRINT CV变量例如距离(DISTANCE),转角(TORSION),回转半径(GYRATION)和坐标数(?COORDINATION)
计算原子组几何中心CENTER
了解如何处理周期性条件,使用WHOLEMOLECULES 和WRAPAROUND,能够确认结果DUMPATOMS.
释放满足特别条件的快照,使用关键字UPDATE_IF.
资源包 在线轨迹包:1
wget https://plumed.github.io/doc-v2.4/user-doc/html/tutorial-resources/trieste-1.tar.gz
文件包含如下内容:
ref.pdb: RNA双链在含有MG离子的水盒子内
traj-whole.xtc: 轨迹文件,周期性处理过后
traj-broken.xtc: 原始GROMACS文件
RNA如下(chimera做图):
PLUMED输入文件例子 一个简单的输入例子:1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
# 告知VIM这是一个plumed格式的文件,若用vim编辑器
# vim: ft=plumed
# 计算原子1和10之间的距离.
# 设置了一个"d"的变量.
d: DISTANCE ATOMS=1,10
# 创建原子20到30之间的中心的虚拟原子位点.
# 设置了一个"center"的变量.
center: CENTER ATOMS=20,30
# 计算 1, 10, 20, 和 center之间的转角.
# 注意的是虚拟位点可以用于真实的原子
# 设置了一个"phi"的变量.
phi: TORSION ATOMS=1,10,20,center
# 计算一些功能函数
# 设置了一个"d2"的变量,计算的为cos phi
d2: MATHEVAL ...
ARG=phi FUNC=cos(x)
PERIODIC=NO
...
# 上面的多行可以写成一行.
# d2: MATHEVAL ARG=phi FUNC=cos(x) PERIODIC=NO
# 打印d,d2,每10步到一个文件命名为 "COLVAR1".
PRINT ARG=d,d2 STRIDE=10 FILE=COLVAR1
# 打印 phi 到另外一个称之为"COLVAR2" 的文件,每 100 步.
PRINT ARG=phi STRIDE=100 FILE=COLVAR2
PS:给的名称例如:d: DISTANCE ATOMS=1,10等价于DISTANCE ATOMS=1,10
然后我们就可以运行进行分析了:
练习1:计算和打印CV 一些原子选择可以通过MOLINFO 关键字来进行_FILL_是需要自己进行修改的:
The gyration radius of the solute RNA molecule (GYRATION ). Look in the ref.pdb file which are the atoms that are part of RNA (search for the first occurrence of a water molecule, residue name SOL). Remember that you don’t need to list all the atoms: instead of ATOMS=1,2,3,4,5 you can write ATOMS=1-5.
The torsional angle (TORSION ) corresponding to the glycosidic chi angle χ of the first nucleotide. Since this nucleotide is a purine (guanine), the proper atoms to compute the torsion are O4’ C1 N9 C4. Find their serial number in the ref.pdb file or learn how to select a special angle reading the MOLINFO documentation.
The total number of contacts (COORDINATION ) between all RNA atoms and all water oxygens. For COORDINATION, set reference distance R_0 to 2.5 A (be careful with units!!). Try to be smart in selecting the water oxygens without listing all of them explicitly.
Same as before but against water hydrogen. Also in this case you should be smart to select water hydrogens. Documentation of GROUP might help.the geometric center of the RNA duplex (use CENTER and DISTANCE ).
需要完整制作的目标如下:
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
# First load information about the molecule.
MOLINFO __FILL__
# Notice that this is special kind of "action" ("setup action")
# that is only used during setup. It will not be re-executed at each step.
# Define some group that will make the rest of the input more readable
# Here are the atoms belonging to RNA.
rna: GROUP ATOMS=1-258
# This is the Mg ion. A group with atom is also useful!
mg: GROUP ATOMS=6580
# This group should contain all the atoms belonging to water molecules.
wat: GROUP ATOMS=__FILL__
# Select water oxygens only:
owat: GROUP __FILL__
# Select water hydrogens only:
hwat: GROUP __FILL__
# Compute gyration radius:
r: GYRATION ATOMS=__FILL__
# Compute the Chi torsional angle:
c: TORSION ATOMS=__FILL__
# Compute coordination of RNA with water oxygens
co: COORDINATION GROUPA=rna GROUPB=owat R_0=__FILL__
# Compute coordination of RNA with water hydrogens
ch: COORDINATION GROUPA=rna GROUPB=hwat __FILL__
# Compute the geometric center of the RNA molecule:
ce: CENTER ATOMS=__FILL__
# Compute the distance between the Mg ion and the RNA center:
d: DISTANCE ATOMS=__FILL__
# Print the collective variables on COLVAR file
# No STRIDE means "print for every step"
PRINT ARG=r,c,co,ch,d FILE=COLVAR
注意,以上的.dat文件中的FILL 需要补全后再运行1
plumed driver --plumed plumed.dat --mf_xtc whole.xtc
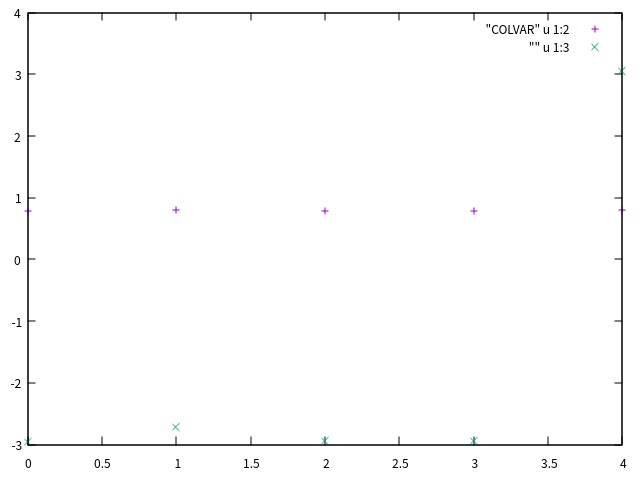
创建的COLVAR文件大致这个样子:1
2
3
4
5
6
7
8
#! FIELDS time r c co ch d
#! SET min_c -pi
#! SET max_c pi
0.000000 0.788694 -2.963150 207.795793 502.027244 0.595611
1.000000 0.804101 -2.717302 208.021688 499.792595 0.951945
2.000000 0.788769 -2.939333 208.347867 500.552127 1.014850
3.000000 0.790232 -2.940726 211.274315 514.749124 1.249502
4.000000 0.796395 3.050949 212.352810 507.892198 2.270682
这个文件可以使用gnuplot查看:1
gnuplot> p "COLVAR" u 1:2, "" u 1:3
以下是我的设置,供参考:1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
# First load information about the molecule.
MOLINFO STRUCTURE=ref.pdb MOLTYPE=rna
# Notice that this is special kind of "action" ("setup action")
# that is only used during setup. It will not be re-executed at each step.
# Define some group that will make the rest of the input more readable
# Here are the atoms belonging to RNA.
rna: GROUP ATOMS=1-258
# This is the Mg ion. A group with atom is also useful!
mg: GROUP ATOMS=6580
# This group should contain all the atoms belonging to water molecules.
wat: GROUP ATOMS=259-6579
# Select water oxygens only:
owat: GROUP ATOMS=wat
# Select water hydrogens only:
hwat: GROUP ATOMS=wat REMOVE=owat
# Compute gyration radius:
r: GYRATION ATOMS=rna
# Compute the Chi torsional angle:
c: TORSION ATOMS=@chi-1
# Compute coordination of RNA with water oxygens
co: COORDINATION GROUPA=rna GROUPB=owat R_0=0.25
# Compute coordination of RNA with water hydrogens
ch: COORDINATION GROUPA=rna GROUPB=hwat R_0=0.25
# Compute the geometric center of the RNA molecule:
ce: CENTER ATOMS=rna
# Compute the distance between the Mg ion and the RNA center:
d: DISTANCE ATOMS=ce,mg
# Print the collective variables on COLVAR file
# No STRIDE means "print for every step"
PRINT ARG=r,c,co,ch,d FILE=COLVAR
我的结果如下:1
2
3
4
5
6
7
8
#! FIELDS time r c co ch d
#! SET min_c -pi
#! SET max_c pi
0.000000 0.788694 -2.963150 206.199511 498.826274 0.595611
1.000000 0.804101 -2.717302 206.427138 496.592883 0.951945
2.000000 0.788769 -2.939333 206.744720 497.333112 1.014850
3.000000 0.790232 -2.940726 209.694868 511.577231 1.249502
4.000000 0.796395 3.050949 210.755463 504.675747 2.270682
两个COORDINATION有点不一样,原因不详
附加:结合多个CV 1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
# Distance between atoms 1 and 2:
d1: DISTANCE ATOMS=1,2
# Distance between atoms 1 and 3:
d2: DISTANCE ATOMS=1,3
# Distance between atoms 1 and 4:
d3: DISTANCE ATOMS=1,4
# Compute the sum of the squares of those three distances:
c: COMBINE ARG=d1,d2,d3 POWERS=2 PERIODIC=NO
# Sort the three distances:
s: SORT ARG=d1,d2,d3
# Notice that SORT creates a compund object with three components:
# s.1: the smallest distance
# s.2: the middle distance
# s.3: the largest distance
p: MATHEVAL ARG=d1,d2,d3 FUNC=x*y*z PERIODIC=NO
# Print the sum of the squares and the largest among the three distances:
PRINT FILE=COLVAR ARG=c,s.3
当有多个距离的时候可以使用部分正则表达式,比如
可以修改为:
MATHEVAL是一个函数功能的实现,非常好用。!
练习1b:结合CV 目的:
The sum of the distances between Mg and each of the phosphorous atoms.
The distance between Mg and the closest phosphorous atom.
要查看亚磷酸原子可以简单的使用shell:1
grep ATOM ref.pdb | grep " P " | awk '{print $2}'
以上脚本主要是用到了管道,grep和awkP(注意空格),最后的行去第二行
练习如下:1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
# First load information about the molecule.
MOLINFO __FILL__
# Define some group that will make the rest of the input more readable
mg: GROUP ATOMS=6580 # a group with one atom is also useful!
# Distances between Mg and phosphorous atoms:
d1: DISTANCE ATOMS=mg,33
d2: DISTANCE __FILL__
__FILL__
d6: DISTANCE __FILL__
# You can use serial numbers, but you might also use MOLINFO strings
# Compute the sum of these distances
c: COMBINE __FILL__
# Compute the distance between Mg and the closest phosphorous atom
s: SORT __FILL__
# Print the requested variables
PRINT FILE=COLVAR __FILL__
结果类似如下:1
2
3
4
5
6
#! FIELDS time c s.1
0.000000 6.655622 0.768704
1.000000 7.264049 0.379416
2.000000 7.876489 0.817820
3.000000 8.230621 0.380191
4.000000 13.708759 2.046935
以下是我的设置,供参考:1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
# First load information about the molecule.
MOLINFO STRUCTURE=ref.pdb MOLTYPE=rna
# Define some group that will make the rest of the input more readable
mg: GROUP ATOMS=6580 # a group with one atom is also useful!
# Distances between Mg and phosphorous atoms:
d1: DISTANCE ATOMS=mg,33
d2: DISTANCE ATOMS=mg,67
d3: DISTANCE ATOMS=mg,101
d4: DISTANCE ATOMS=mg,165
d5: DISTANCE ATOMS=mg,196
d6: DISTANCE ATOMS=mg,227
# You can use serial numbers, but you might also use MOLINFO strings
# Compute the sum of these distances
c: COMBINE ARG=(d.) PERIODIC=NO
# Compute the distance between Mg and the closest phosphorous atom
s: SORT ARG=(d.)
# Print the requested variables
PRINT FILE=COLVAR ARG=c,s.1
我的结果如下:1
2
3
4
5
6
#! FIELDS time c s.1
0.000000 6.655622 0.768704
1.000000 7.264049 0.379416
2.000000 7.876489 0.817820
3.000000 8.230621 0.380191
4.000000 13.708759 2.046935
解决周期性问题 PLUMED可以使用DUMPATOMS来转存(dump)内部储存的原子坐标,主要可能用到的用途如下:
To dump coordinates of virtual atoms that only exist within PLUMED (e.g. a CENTER).
To dump snapshots of our molecule conditioned to some value of some collective variable (see UPDATE_IF).
To dump coordinates of atoms that have been moved by PLUMED.
我们可以用VMD来查看GROMACS未处理周期性的轨迹(其实我感觉处理周期性完全可以在GROMACS中处理后交由PLUMED完成后续分析)
1
vmd ref.pdb traj-broken.xtc
练习2:解决周期性问题并且转存原子坐标 要求如下:
保证RNA完整,详细参考WHOLEMOLECULES
提供一个模板进行比对(structure reference.pdb).具体查看FIT_TO_TEMPLATE ,使用TYPE=OPTIMAL.如若使用ref.pdb,需要移除非 RNA的原子
完整的RNA和Mg离子,但是包括的快照为Mg和残基8的亚磷酸(P)小于4A的距离. 使用UPDATE_IF 选择执行框架
整个RNA双链体加上水分子和mg离子缠绕在双链体的中心。 先用CENTER计算双螺旋的中心,然后用WRAPAROUND 包裹分子。 确保移动后个别水分子不会破碎!
练习需要填充的配置文件如下:1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
# First load information about the molecule.
MOLINFO __FILL__
# Define here the groups that you need.
# Same as in the previous exercise.
rna: GROUP ATOMS=__FILL__
mg: GROUP ATOMS=__FILL__
wat: GROUP ATOMS=__FILL__
# Make RNA duplex whole.
WHOLEMOLECULES __FILL__
# Dump first trajectory in gro format.
# Notice that PLUMED understands the format based on the file extension
DUMPATOMS ATOMS=rna FILE=rna-whole.gro
# Align RNA duplex to a reference structure
# This should not be the ref.pdb file but a new file with only RNA atoms.
FIT_TO_TEMPLATE REFERENCE=__FILL__ TYPE=OPTIMAL
# Notice that before using FIT_TO_TEMPLATE we used WHOLEMOLECULES to make RNA whole
# This is necessary otherwise you would align a broken molecule!
# Dump the aligned RNA on a separate file
DUMPATOMS ATOMS=rna FILE=rna-aligned.gro
# Compute the distance between the Mg and the Phosphorous from residue 8
d: DISTANCE ATOMS=mg,__FILL__ ## put the serial number of the correct phosphorous here
# here we only dump frames conditioned to the value of d
UPDATE_IF ARG=d __FILL__
DUMPATOMS ATOMS=rna,mg FILE=rna-select.gro
UPDATE_IF ARG=d __FILL__ # this command is required to close the UPDATE_IF above
# compute the center of the RNA molecule
center: CENTER ATOMS=rna
# Wrap atoms correctly
WRAPAROUND ATOMS=mg AROUND=__FILL__
WRAPAROUND ATOMS=wat AROUND=center __FILL__ # anything missing here?
# Dump the last trajectory
DUMPATOMS ATOMS=rna,wat,mg FILE=rna-wrap.gro
我的设置与结果1
grep -v "SOL" ref.pdb | grep -v "MG" > refrna.pdb
我的配置如下:1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
# First load information about the molecule.
MOLINFO STRUCTURE=ref.pdb MOLTYPE=rna
# Define here the groups that you need.
# Same as in the previous exercise.
rna: GROUP ATOMS=1-258
mg: GROUP ATOMS=6580
wat: GROUP ATOMS=259-6579
# Make RNA duplex whole.
WHOLEMOLECULES ENTITY0=1-258
# Dump first trajectory in gro format.
# Notice that PLUMED understands the format based on the file extension
DUMPATOMS ATOMS=rna FILE=rna-whole.gro
# Align RNA duplex to a reference structure
# This should not be the ref.pdb file but a new file with only RNA atoms.
FIT_TO_TEMPLATE REFERENCE=refrna.pdb TYPE=SIMPLE
# Notice that before using FIT_TO_TEMPLATE we used WHOLEMOLECULES to make RNA whole
# This is necessary otherwise you would align a broken molecule!
# Dump the aligned RNA on a separate file
DUMPATOMS ATOMS=rna FILE=rna-aligned.gro
# Compute the distance between the Mg and the Phosphorous from residue 8
d: DISTANCE ATOMS=mg,227 ## put the serial number of the correct phosphorous here
# here we only dump frames conditioned to the value of d
UPDATE_IF ARG=d LESS_THAN=0.5
DUMPATOMS ATOMS=rna,mg FILE=rna-select.gro
UPDATE_IF ARG=d END # this command is required to close the UPDATE_IF above
# compute the center of the RNA molecule
center: CENTER ATOMS=rna
# Wrap atoms correctly
WRAPAROUND ATOMS=mg AROUND=rna
WRAPAROUND ATOMS=wat AROUND=center GROUPBY=3 # anything missing here?
# Dump the last trajectory
DUMPATOMS ATOMS=rna,wat,mg FILE=rna-wrap.gro
执行命令:1
plumed driver --plumed plumed.dat --mf_xtc broken.xtc
我使用的是TYPE=SIMPLE ,但是教程上建议的是 OPTIMAL ,但是我一使用就报错,暂时不知道原因,也不知道自己哪里错了,不知道是不是bug
参考视频:练习1b 练习1