安装教程
之前了解了以下Epock,前一篇文章也使用了Epock做分析,最大的好处就是简便,但是其分析是建立与自己处理口袋上的,这样误差就非常的大,导致模拟往往开始的时候体积高,后期下滑的很厉害~看到一篇plos one 上使用了fpocket2,所以使用了解一下。fpocket2有一个好处是有在线服务:点我进去
要求:基于可视化软件VMD和Pymol
安装:
|
|
如果遇到错误,将makefile中部分内容进行替换:
|
|
fpocket基本使用
软件自带了一个sample的文件夹,里面有许多例子,我们可以使用一个最为简单的
fpocket -f sample/3LKF.pdb
终端会有如下:
=========== Pocket hunting begins ==========
=========== Pocket hunting ends ============
表示已经使用运算完毕
会有一个3LKF_out的文件夹,可以使用3LKF_VMD.sh脚本快速查看
结果如下:
[](http://kangsgo.com/wp-content/uploads/2016/09/3LKF.png)
修改球的大小颜色位于3LKF_out处的Graphical->Representations
[](http://kangsgo.com/wp-content/uploads/2016/09/1.png)
同样可以运行pymol脚本3LKF_PYMOL.sh
和手册上的图感觉不同,还需要进一步微调
[](http://kangsgo.com/wp-content/uploads/2016/09/2.png)
dpocket口袋特征提取
dpocket的d一词为describing的意思,能够描述口袋的物理化学环境,并且能够分析多个结构。
例子:
数据要求设计一个text文件,该文件包含蛋白的pdb结构文件,以及配体的ID,教程上说位于sample/test_dpocket.txt上,但是我并未看见,而是有一个dpinput.txt文件,估计是1.0版本遗留下来的产物,我们重命名并且修改中间的内容变为如下:(!注意pdb与配体名称中间为制表符【table】,7TTA的结构文件可能需要自己下载!)
|
|
dpocket -f sample/test_dpocket.txt
会得到3个文件:
|
|
可以使用-o 来修改输出文件前缀,如-o my_test则为my_test_explicitp.txt
fpocketp.txt描述精确的口袋的相关描述
fpocketnp.txt为描述非精确的口袋
explicito.txt则为描述自定义参考残基与配体相互之间距离多少被纳入分析范围(默认4A)
得到的ASCII 文本文件可以用统计学软件例如R进行分析(自己待研究)
tpocket评分功能
相比fpoket与dpocket,其是成药性预测评分的进一步精细预测,T为Test的含义,测试你需要比对有无配体的结构在口袋上的空间对应关系。
例子:tpocket对于无配体结构
这个需要下载fpocket-1.0-date
需要有pp_apo-t.txt 和 pp_cplx-t.txt 两个文件。前者为无配体,后者为包含配体,pp_apo-t.txt 部分示例如下:
|
|
运行:
|
|
运行需要花费一些时间,最后可以得到两个文件,第一个为默认的stats_g.txt 他包含了fpocket所需要的所有的指标
|
|
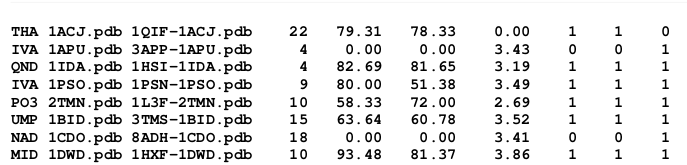
此结果为48个蛋白的总计stats_p.txt为分开的信息
部分如下:
若为计算有配体的结构,则只需要把无配体结构也改为有配体即可如:
|
|
mdpocket MD轨迹中的口袋检测
mdpocket主要有如下功能:
1.MD轨迹中的口袋检测
2.口袋瞬时可视化
3.MD轨迹中口袋描述的提取
4.MD轨迹中获取口袋事件的静态图像
使用的格式为pdb格式
例子:
非常重要的是首先需要align所有的snapshots
以Amber为例:
1.创建ptraj输入文件,包含以下内容
|
|
2.运行ptraj使用如下命令
|
|
主要方法内容为切割为3个文件,每个为250 snaphots,取第10个计算。设置参考的PDB结构进行校准。strip用来丢弃除去蛋白的一切东西,如溶剂,离子等(注:因为这个蛋白只有208位,所以1-208,貌似只会获取残基,未考)接下来,我们比对每个snapshot和参考结构,仅使用重原子残基 25-88和120-196.输出到snapshots/snap.pdb,名称为snap.pdb.#,你需要重命名你的pdb文件成为例如snap_#.pdb。可以使用如下shell命令:
|
|
最后你需要对你的snapshots文件进行排序,可以使用scripts目录中的createMDPocketInputFile.py,命令如下:
|
|
将会输出一个mdpocket_input.txt文件
|
|
最后会获得如下文件:
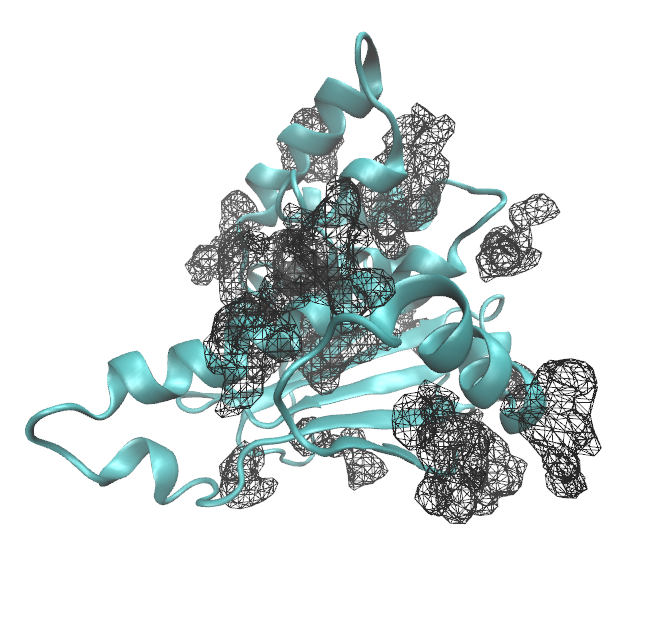
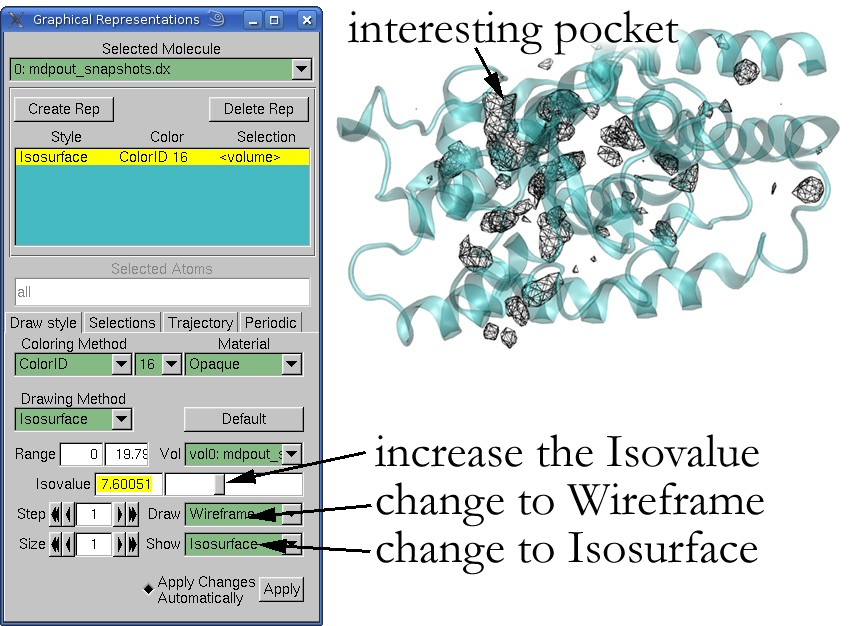
mdpout_dens_grid.dx 其会显示体积坐标,设置和最终效果图如下:
mdpout_freq_grid.dx 这个文件和前者非常类似,但是这个网格只包含在MD轨迹中开放频率的量度
mdpout_dens_iso_8.pdb 这个文件包含在8A^3大于3或者更大的Voronoi Vertices, Pymol可以选择并存储你感兴趣的格点,一般情况下蛋白结合口袋或者蛋白蛋白作用口袋选择isovalue高的,例如5.若是研究构象的改变例如开关闭合,则选择2或者3这种isovalue较低的。可以参考高级功能参看如何提取。
mdpout_freq_iso_0_5.pdb 与前者相似,只是cut -off 为0.5
这样即可使用自己选择的结合口袋进行计算:
|
|
-v 是一个可选选项,用以控制合理的计算时间(我认为是,若没有计算完,也会停止?)将会获得如下文件:
mdpout_mdpocket.pdb 所选择的口袋区域,仅可使用PyMOL读取。
mdpout_mdpocket_atoms.pdb 相互作用原子
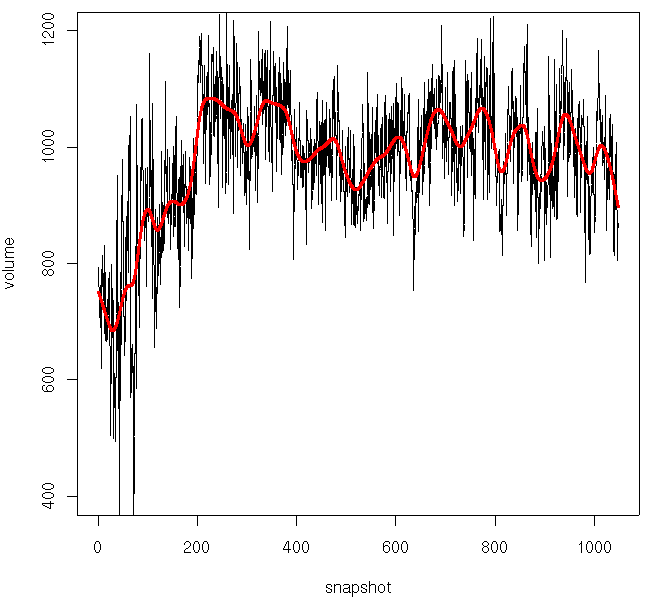
mdpout_descritpors.txt 体积等信息
可以使用R等作图
|
|
如下: