SMINA教程以CDPK1为例
原作者:David Ryan Koes
翻译:康文渊
若进行精读建议查看原文以及参考文献,本文是我根据教程快速阅读做的笔记
Q1:我们有autodock vina,为什么还要SMINA?
A1:SMINA有更加优秀的速度和更加的扩展性
Q2:用该对接软件有什么优势?
A2:首先该对接软件为免费/学术免费软件,其次其有非常良好的对接准确度,甚至在商业软件中都为上乘,同时可以很好的运用SMINA进行得分改造,进一步提升性能和速度
本教程是一个使用SMINA进行对E.tenella中的钙依赖性蛋白激酶1(calcium-dependent protein kinase 1 CDPK1)进行的结构为基础的虚拟筛选的教程。E.tenella是一种感染幼禽并且导致潜在致命球虫病的寄生虫。尽管这种酶并未解析晶体结构(译者翻译时该结构已经被解析,pdb:4YSJ),但是有相关寄生虫的该种酶的结构。C. parvum 和 T. gondii的该种酶结构已经被解析,并且有生物活性数据表明他们的小分子竭抗剂。
我们将会对怎么分析已知结构,评价SMINA对于这些结构的表现,对于对接结果创建自定义的得分函数,使用SWISS-MODEL创建E.tenella的结构模型,并且进行E.tenellaCDPK1的虚拟筛选。(个人认为作者提到的E.tenella均表示的为E.tenellaCDPK1)。
假设和惯例
假设读者对Linux命令行有一定的了解,并对一些用到的软件熟悉。教程在Ubuntu Linux12.04(原作者)和Ubuntu Linux16.04(译者)测试通过。bash命令行如下:
python基础的PyMOL环境展示如下:
以下为(译者)用到的软件:
|–Software–|–License–|–Source–|
|—|—|—|
|color_by_mutation|GNU|http://www.pymolwiki.org/index.php/Color_By_Mutations|
|PyMOL 1.7.x|Python|http://sourceforge.net/projects/pymol/|
|RDKit 2017_03|BSD Style|http://rdkit.org/|
|smina|GPLv2|http://sourceforge.net/projects/smina/|
1. 检查C.parvum和T.gondii晶体结构
该部分内容可以参考早期博客文章同源建模系列
首先我们分析E.tenella序列和验证三个结构的异同(三个寄生虫中改结构)。然后我们使用C.parvum和T.gondii创建一个虚拟筛选的测试集,并且对接这些测试集化合物。我们使用这些对接结果来进行晶体结构的筛选和模型的增加。
1.1 靶标分析
首先,我们分析E.tenella序列和C.parvum和T.gondii*结构
1.1.1 E.tenella序列分析
1.复制或者下载E.tenella的FASTA序列,你可以点击这里进入网址,同样也可以下载我们已经下载好的。
2.进行BLAST搜索,若你为进入的网址,可以直接点击ncbi右侧的BLAST进行搜寻(如下图)。若为下载的文件可以进入BLAST页面上传fasta文件进行搜寻。
3.进入页面。
4.点击BLAST。
5.结果如图3,可以发现得分最高的为已经解析的结构,相似度为99%,但是我们在这里假装没有。排第二的为Neospora Caninum(4M97),但是更加相近的为T.gondii的几个结构(4M7N,3KU2,3I79,3HX4,3I7C),相似度大于80%。C.parvum的首次命中(3IGO)相似度为62%。
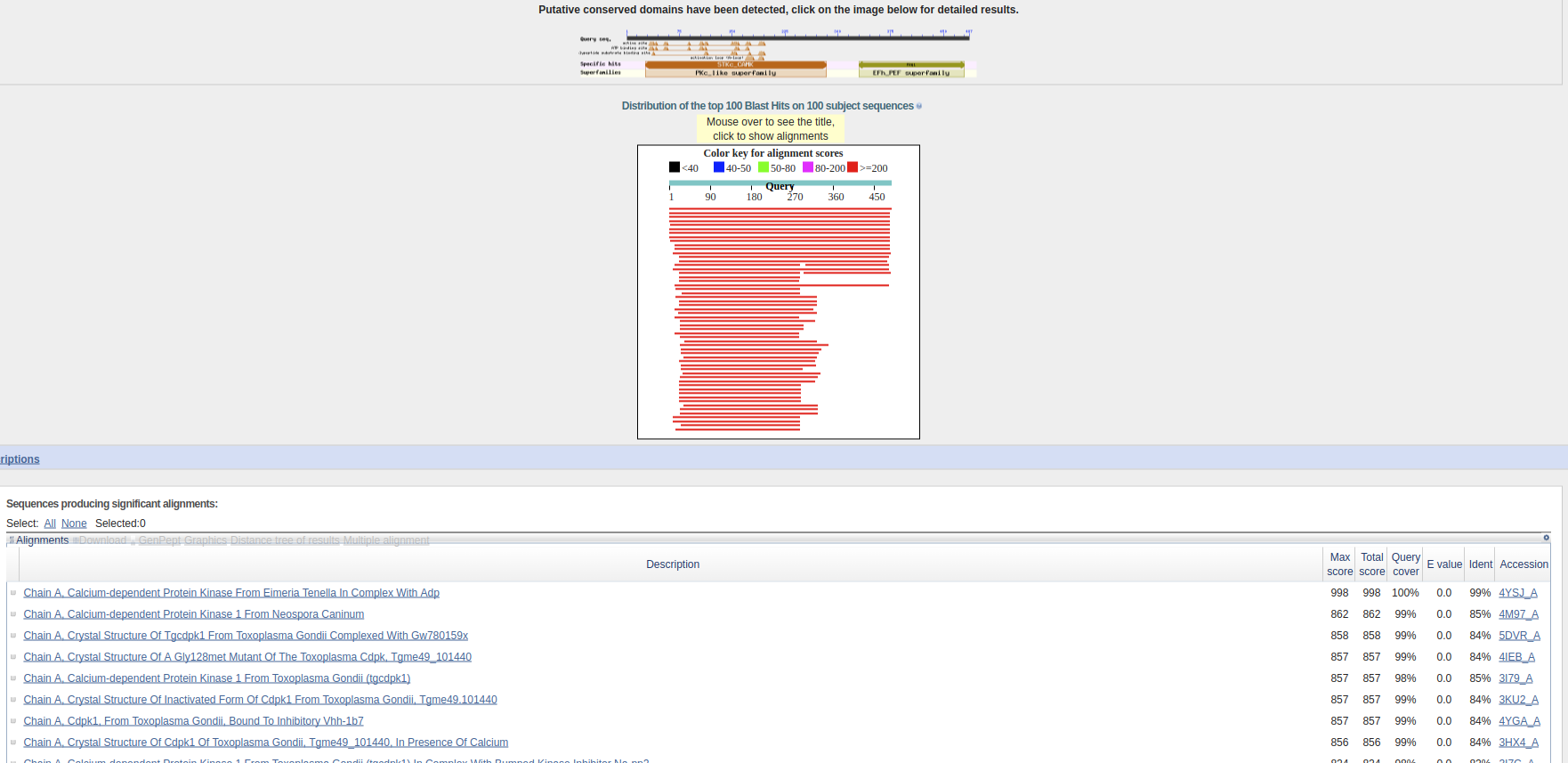
从中我们得出的结论为T.gondii的CDPK1相比于C.parvum具有更高的相似度。建议T.gondii结构选为同源建模的模板更为合理。
1.1.2 结构分析
为了鉴定T.gondii和E.tenella之间CDPK1结合位点的差别,我们创建了一个E.tenella同源模型。并且使用color_by_mutation和PyMOL进行结构可视化序列的不同。
1.在SWISS-MODEL进行自动建模,你也可以参考之前的教程
译者注:我认为该方法并不是建模的最佳方法,最佳方法为找到具有共同抑制剂的晶体结构进行基于配体的多模板建模,这样可以保证重要的活性腔的相似,而不是使用没有结合配体的晶体结构进行建模。
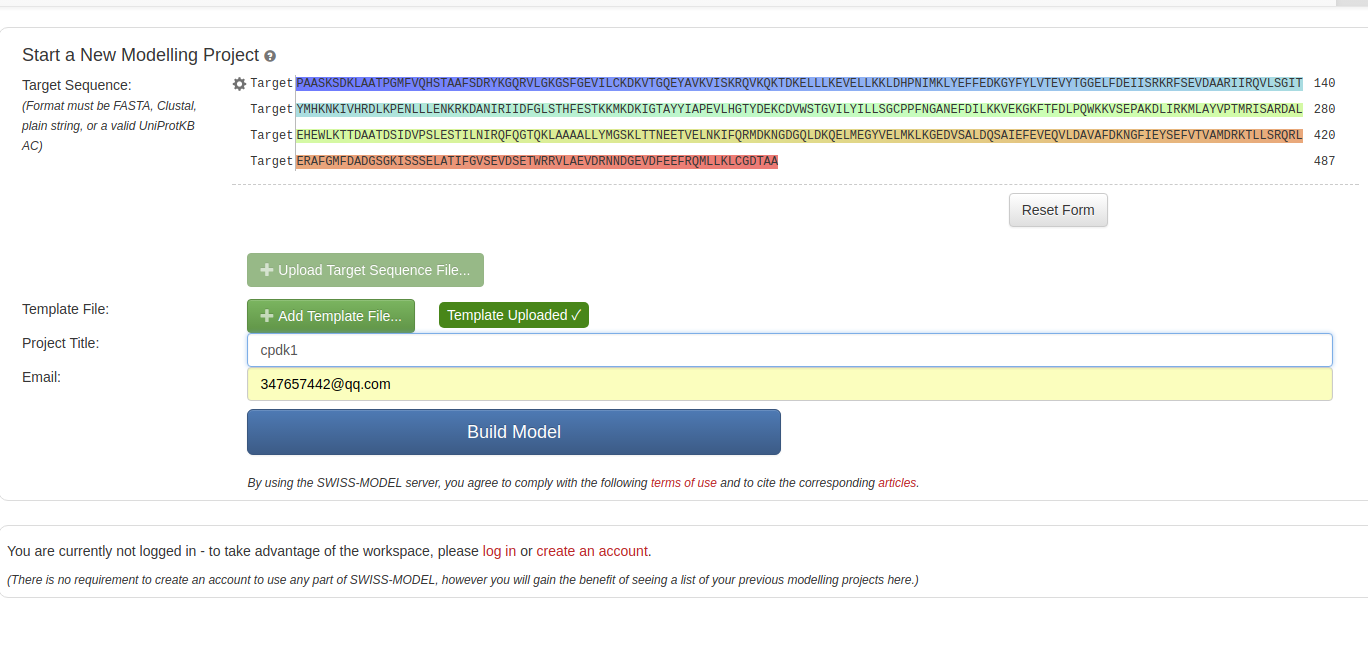
2.输入E.tenella的FASTA序列和3I79进行建模(图4)
3.下载结果文件,你可以在这里下载我已经建模好的。
4.在PyMOL中加载模型,3I79和3NCG
|
|
译者注:在这里
3NCG来的有点突然,搜索一下可以发现3NCG为C.parvum的激活状态下的晶体结构
5.比对结构
|
|
6.从3NCG中提取BK1配体,并且隐藏该pdb的其余结构,因为3I79为非结合状态,我们使用这个配体来占有结合位点
|
|
7.探索模型和3I79结合位点周围的残基,值得注意的是SWISS-MODEL忠实的复制了侧链构像。
8.着色两者之间的突变位点
|
|
9.探索结合位点,注意的是3I79中GLY128在E.tenella模型中被突变为THR 108,其余在结合位点是大体不变的。
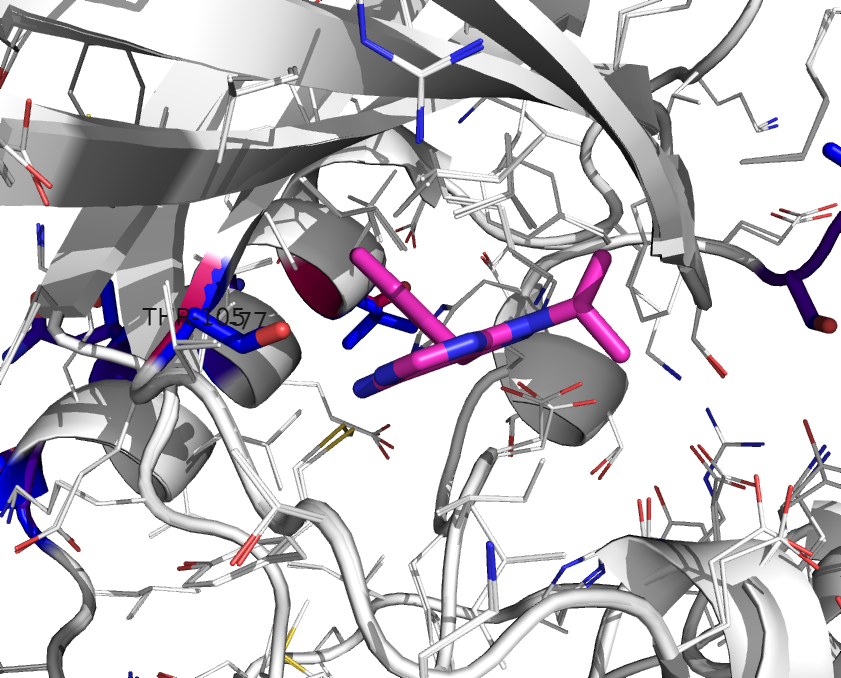
10.重复分析3NCG,可以发现C.parvum在结合位点附近具有更多的突变
1.2 测试集的创建
PubChem上有T.gondii和C.parvum的CDPK1诘抗击。我们使用这些数据作为虚拟筛选的测试集对这两个靶标进行筛选.
1.2.1组装活性化合物
(a)在PubChem搜索CDPK1然后排序活性化合物,每个靶点选择三个最大的化合物数据集,为了保证和教程一致,我们选择的为和原教程相同的数据集.
T.gondii
aid:677062,600378,672956
C.parvum
aid:600379,672955,66097
(b)选择active,然后点击Structures下载,之后选择没有压缩的SMILES格式,可以点此下载我们下载好的
(c)修正SMILES结构。下载的SMILES文件字符串和标题顺序相反。另外,我们需要去除salt,并将关键字“active”添加到所有活性化合物的标题中。我们每个文献执行如下命令(例如aid677062.txt为例):
awk主要是进行了前后的颠倒,并且在标题后增加active标签,sed主要是将Cl进行替换,替换成空,中间用管道链接,输出到smi文件格式。
(d)将每个靶点的内容合并为单个文件
|
|
T.gondii和C.pavum分别包含156个和89个活性化合物。你可以在这里下载合并好的文件。
1.2.2 创建诱饵数据集
该在线网站说只需要10min,实际上需要1-2天左右时间
由于这些靶点的无活性配体数量较少,我们使用诱饵数据库Database of Useful Decoys:Enhanced(DUDE)方法来从ZINC数据库中进行采样。这些诱饵被选择的方法为化学性质不相同(即和有活性的配体相比较不相似,认为是不结合的)。但分子质量,logP,旋转键和氢键受体供体这些简单的分子参数是相同的。
(a)使用DUDE网站创建诱饵数据集,将会生成dude-decoys.tar.gz文件,里面包含50个参数匹配的诱饵。
(b)将诱饵结合进入单个文件
|
|
点此下载decoys文件,(T.gondii诱饵和活性化合物。C.pavum诱饵和活性化合物)。
1.2.3 生成三维结构
我们将使用开源软件RDKit利用2D SMILES生成3D构像
(a)将RDKit写入你的PYTHONPATH,并且执行rdconf.py脚本
|
|
(b)每个active/decoy生成单一构像
|
|
(c)将文件合并
|
|
由于有两个文件,我们将T.gondii和C.pavum分别命令为gondii.sdf和parvum.sdf
1.3 对接
我们使用SMINA通过对接化合物进入受体结构进行虚拟筛选,采用AutoDock Vina得分功能。由于我们对一个固定的受体对接,所以我们选择一个好的受体结构是非常重要的。我们将会使用CDPK1的测试集来模拟评价我们的对接工具和选择受体结构。
1.鉴定PDB中所有C.parvum和T.gondii文件,搜索’calcium-dependent protein kinase 1’,并且选择恰当的生物(organism.)
C. parvum: 2QG5 2WEI 3DFA 3F3Z 3HKO 3IGO 3L19 3LIJ 3MWU 3NCG
T. gondii: 3I79 3I7B 3I7C 3KU2 3N51 3NYV 3SX9 3SXF 3T3U 3T3V 3UPX 3UPZ 3V51 3V5P 3V5T 4M84
由于教程较老,故和自己搜索有差异
2.下载这些pdb文件,你可以用如下的简单方法,或者直接下载我下载好的。
将以下代码保存为脚本,例如down.sh
执行如下命令:
|
|
3.比对和提取结构。在pymol中打开靶标pdb文件。比对他们。除去水和原子。提取每个配体到自己的对象。
|
|
4.保存受体和配体文件,注意的是3LI9没有酶结构域,3DFA和2QG5为未结合态结构,综合考虑我们将其删除。
|
|
保存(注意一下需要保存为python文件后run进行操作)
|
|
5.将配体导入进单一文件,由于我们之前做了,该步骤无需做
6.对接测试集化合物到每个受体结构,例如:
|
|
在这里我们制定了一个随机种子。结合盒子采用autobox_ligand设置,创建的盒子大小为对于提供的配体8埃缓冲。配体,受体和输出文件可以使用任意OpenBabel支持的格式。
7.获取每个化合物输出得分最高的对接姿势。
|
|
8.分析对接结果,原文是使用的R进行的分析,大家可以以下做参考,当然也可以依葫芦画瓢使用Python进行处理。
(a)R版本方法如下:
|
|
图大致如下:
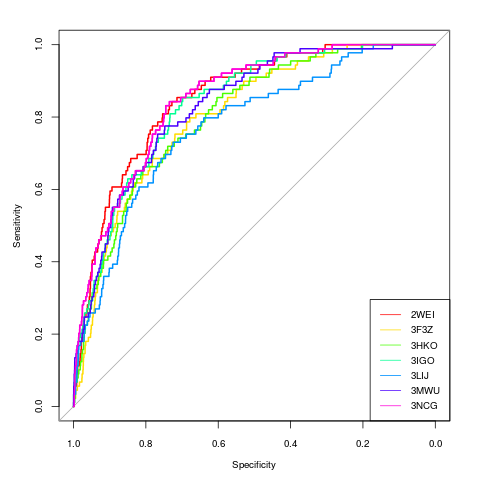
AUC和Partial AUC的值分别为0.8520,0.6485。此数据运用的为官方数据,python版本为我自己写的方法,供参考
(b) ython版本如下:
首先我们使用awk处理一下原始文本
|
|
得到的文件可以从这里下载,python脚本如下:
|
|
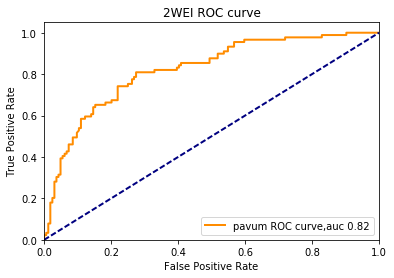
2. E.tenella CDPK1 模型和 测试集预测
现在我们创建一个E.tenella模型来进行虚拟筛选和发展基于先前得分结果的用户自定义的得分功能。
2.1 建模
基于之前章节的对接结果,T.gondii在全局和结合位点的序列中是更加接近E.tenella的.3T3U是T.gondii最优得分性能的一个结构(AUC和早期经验性AUC评价),因此我们使用SWISS-Model构建swissmodel3T3U.pdb
2.2 对接测试集
1.转化为SMILES格式
|
|
2.生成构像
|
|
3.对接测试集
|
|
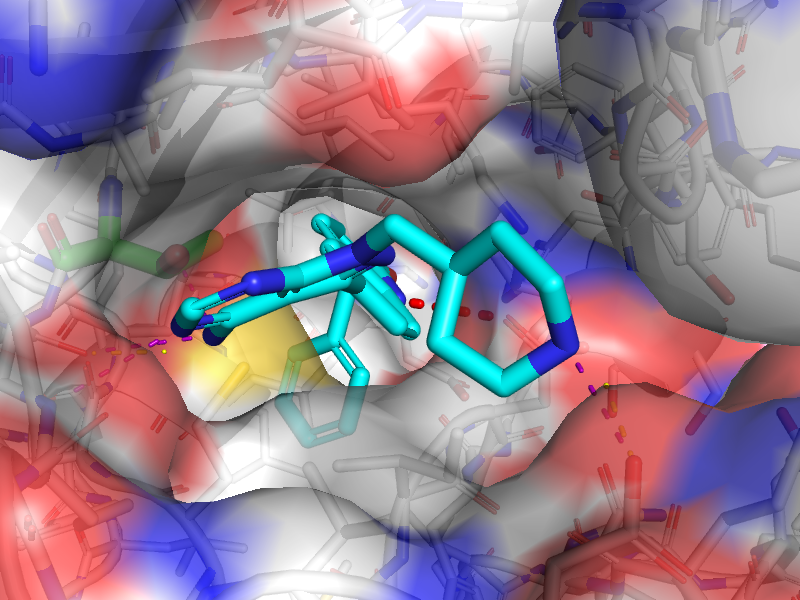
4.选择和排序得分
|
|
最佳得分构像如下图
2.3 用户自定义得分
我们发展了用户自定义得分功能用以T.gondii测试集参数化。增加到默认的得分功能上。
1.删除atom-types项目(如果考虑所有可能的原子类型组合,这将是一个非常大的项目),同样包括默认的第二Gaussian项目,他同样存在于默认得分之中。所有的权重均设为1.0
|
|
grep -v -E的用法比较少,-v表示不包含匹配文本的所有行,-E为启用正则表达式
2.得分最高的构像3T3U测试集拿来进行比较,由于我这里只做了2WEI,所以实际上此步我用的2WEI
|
|
原文档中最后一行为/active/,但是个人觉得并没有达到区分active和unactive的用途,故本人对其进行了修改。可以从此下载文件。
3.作者使用R中的rms包和logistical回归来进行后向变量选择来契合活性数据的得分。为了防止过拟合,只选择p-值较低的功能(<.0001)。
|
|
4.保存测定的系数coefficients以创建新的评分函数。因为项目名称中包含特征符,其在R中是无效命名,所以需要编辑输入以重新储存适当的terms,得分函数的结果如图下:

5.通过将系数乘以每一项的平均值来创建该评分函数中每个项的贡献的平均估计
|
|
结果如下图

疏水性,VDW,氢键和溶剂化为有利相互作用,非疏水性,排斥性和受体-受体为负相关作用。VDW和gauss具有非常强的权重,线性氢键和Lennard Jones氢键展示了一些权重。
4*.Python包实现,Python的scikit-learn中对于logistical并没有P-值,译者对与R也不是特别理解,不知道能否领会作者的含义,以下仅供参考。 我们采用scikit-learn包中的logistical回归,同时将数据为训练集(75%)与测试集,检测是否过拟合,若没有测试集效果也不错,就使用所有的作为训练集再次训练。
|
|
结果如下:
译者计算出来和原作者计算出来有差异,主要就是hydrophobic和non_hydrophobic项两者的权重都为负数,项和值上也有差异。接下来我们乘上平均值得到最终权重。
|
|
结果如下:
6.评价这个新的得分函数,使用这个新的得分函数fitscore(个人觉得是第一次得出的权重进行的计算,因为我用第二次的权重进行计算数值很大,此处待考)除了重新打分最高的对接姿势以外,我们重新打分和重新排序所有的对接姿势。值得注意的是这个得分函数是预测的活性而不是亲和力。越大的positive值表示的是复合物可能具有活性的概率更大,所以复合物在选择最佳构像排序的时候需要逆转数字顺序。
|
|
译者在做的时候对第6条不是很理解
7.使用新的得分函数对接E.tenella化合物集:
|
|
应该使用在这个测试集上表现更好的评分函数来进行完整的虚拟筛选选择化合物,结果可以从此下载
译者认为自定义得分并未达到非常好的效果,可以进一步优化
3.虚拟筛选
已经阐明精心设计的自定义得分可以很好的应用在E.tenella结构模型,我们进行筛选eMolecules化合物库。需要注意的是我们的自定义得分函数并不使用于对接,而是参数化预测已经对接好的姿势的活性。
1.下载eMolecules数据库
|
|
2.根据环境切割不同大小。例如,10000每块进行切割
|
|
3.生成构像
|
|
4.对接每个块
|
|
5.对获得的最高排名姿势使用默认的得分和自定义得分进行评价
|
|
6.将对接姿势放入一个文件并排序
|
|
参考文献:
Ojo KK, Larson ET, Keyloun KR, Castaneda LJ, Derocher AE, Inampudi KK, Kim JE, Arakaki TL, Murphy RC, Zhang L, Napuli AJ, Maly DJ, Verlinde CL, Buckner FS, Parsons M, Hol WG, Merritt EA, Van Voorhis WC. (2010) Toxoplasma gondii calcium-dependent protein kinase 1 is a target for selective kinase inhibitors. Nat Struct Mol Biol. 17:602-7. PMID: 20436472 PMCID: PMC2896873
Murphy RC, Ojo KK, Larson ET, Castellanos-Gonzalez A, Perera BG, Keyloun KR, Kim JE, Bhandari JG, Muller NR, Verlinde CL, White AC, Merritt EA, Van Voorhis WC, Maly DJ. (2010) Discovery of Potent and Selective Inhibitors of Calcium-Dependent Protein Kinase 1 (CDPK1) from C. parvum and T. gondii. (2010) ACS Med Chem Lett. 1(7):331-335. PMID: 21116453 PMCID: PMC2992447
Johnson SM, Murphy RC, Geiger JA, Derocher A, Zhang Z, Ojo K, Larson E, Perera BG, Dale E, He P, Fox A, Mueller N, Merritt EA, Fan E, Reid M, Parsons M, Van Voorhis WC, Maly DJ. (2012) Development of Toxoplasma gondii Calcium-Dependent Protein Kinase 1 (TgCDPK1) Inhibitors with Potent Anti-Toxoplasma Activity. J Med Chem. 55(5):2416-26. PMID: 22320388 PMCID: PMC3306180
Larson ET, Ojo KK, Murphy RC, Johnson SM, Zhang Z, Kim JE, Leibly DJ, Fox AM, Reid MC, Dale EJ, Perera BG, Kim J, Hewitt SN, Hol WG, Verlinde CL, Fan E, Van Voorhis WC, Maly DJ, Merritt EA. (2012) Multiple Determinants for Selective Inhibition of Apicomplexan Calcium-Dependent Protein Kinase CDPK1. J Med Chem. 55(6):2803-10. PMID: 22369268 PMCID: PMC3336864
Ojo KK, Pfander C, Mueller NR, Burstroem C, Larson ET, Bryan CM, Fox AMW, Reid MC, Johnson SM, Murphy RC, Kennedy M, Henning Mann H, Leibly DJ, Hewitt SN, Verlinde CLMJ. Kappe S, Merritt EA, Maly DJ, Billker O, Van Voorhis WC. (2012) Transmission of malaria to mosquitoes blocked by bumped kinase inhibitors. J. Clin. Invest. 122(6):2301–2305. doi: 10.1172/JCI61822 PMID:22565309 PMCID: PMC3366411
Zhang Z, Ojo KK, Johnson SM, Larson ET, He P, Geiger JA, Castellanos-Gonzalez A, White AC Jr, Parsons M, Merritt EA, Maly DJ, Verlinde CL, Van Voorhis WC, Fan E. (2012) Benzoylbenzimidazole-based selective inhibitors targeting Cryptosporidium parvum and Toxoplasma gondii calcium-dependent protein kinase-1. Bioorg Med Chem Lett. 22:5264-7. PMID: 22795629 PMCID: PMC3420979
Castellanos-Gonzalez A, A. White AC Jr, Ojo KK, Vidadala R, Zhang Z, Reid MC, Fox AMW, Keyloun KR, Rivas K, Irani A, Dann SM, Fan E, Maly DJ, Wesley C Van Voorhis. (2013). A novel Calcium Dependent Protein Kinase Inhibitor as a lead compound for treating Cryptosporidiosis. J Infect. Dis. PMID: 23878324 2013 Jul 21. [Epub ahead of print]
Ojo KK, Eastman RT, Vidadala R, Zhang Z, Rivas KL, Choi R, Lutz JD, Reid MC, Fox AMW, Hulverson MA, Kennedy M, Isoherranen N, Kim LM, Comess KM, Kempf DJ, Verlinde CLMJ, Su X-Z, Kappe S, Maly DJ, Fan E, & Van Voorhis WC. (2013) Specific inhibitor of PfCDPK4 blocks malaria transmission: Chemical-genetic validation. J Infect Dis. 2013 Oct 10. [Epub ahead of print] PMID: 24123773

