阅读前请注意:
1.此文仅是我看Martini文献的一些总结,若需要使用Martini力场建议仔细的查看官网信息和相关文献。此文花费较大精力,若有什么意见或者建议欢迎发邮箱交流。邮箱为kangsgo@vip.qq.com 湖大-小康
2.由于我只关注蛋白模拟,故只查阅了Martini力场2.0的文献以及蛋白参数文献
3.此笔记并未包含Martini力场性能测评部分,可能后续会加上
粗粒化(CG )模拟在各种各样的模拟化技术中提供与全原子模型(AA model)相比更广的空间和时间尺度研究。在粗粒化模拟研究的早期一般运用在蒙特卡洛模拟上,这是一种基于概率的模拟算法,与经典模拟在理论上还是有大的差别,早期的CG模型其缺点是需要每次模拟前都进行参数的设置矫正,而Martini力场开发的初衷是更为提供一种能够进行普适应用的操作简单的力场。
Martini的力场开发目的主要是应用于囊泡的形成,薄层相的转移,结构和膜的自组装等等,其比较关注于极性与非极性之间的相互转换/作用。Martini力场相比之前的力场模拟(2007年以前)有更多的能量”等级”和类型,与实验参数也拟合的更好,同时提供了环类物质的CG 模型的方法。
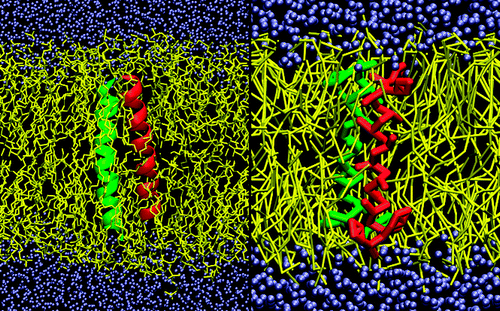
力场概述(2.0版本):
联系位点:
Maritini力场与其他CG力场一样,均为4个heavy原子用一个单一珠子所联系。珠子主要有四种主要类型。polar(P),nonpolar(N),apolar(C)和charged(Q)。同时有18种亚型,比如按照成氢键能力用单字母分为(d=donor,a=acceptor,da=both,0=none)或者指示极性(1,最低极性,5,最高极性)
蛋白的原子类型:
蛋白残基中侧链表示:非极性残基Leu,Pro,Ile,Val,Cys和Met用C-类型颗粒表示。非极性残基Thr,Ser,Asn和Gln通过P-类型颗粒表示,氨基酸残基带有小的负电荷侧链Glu和Asp残基用Q-类型颗粒表示。正电荷氨基酸残基Arg和Lys采用带电荷的Q类型和不带电荷颗粒组成。带环芳香烃残基通过三(His,Phe和Tyr)或者四(Trp)个珠子来表示特别的环颗粒。Gly和Ala残基仅仅设置骨架颗粒。骨架参数依赖二级结构,如下图:
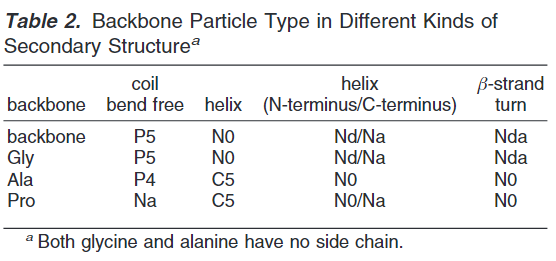
在溶液中或者无规卷曲或者弯曲中,骨架拥有非常强的极性特征(P类型),在部分螺旋或者β折叠中,内部骨架键减少了极性特征的放大(N类型)。辅氨酸是低极性的因为其缺乏氢键供体能力。其全原子映射结果如下表:
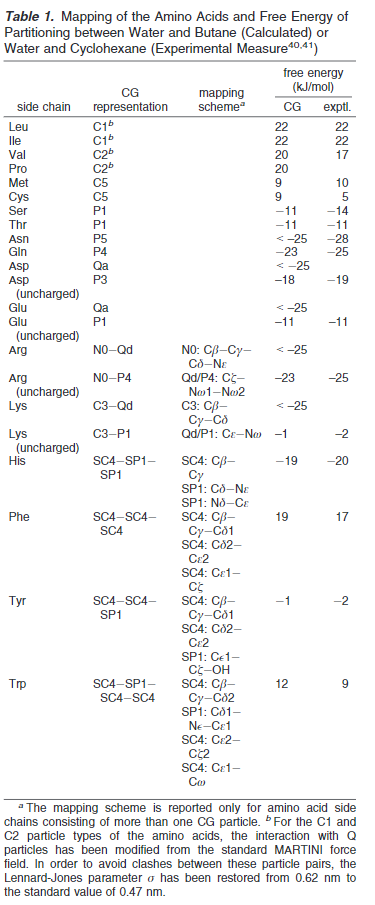
非键联系:
首先是Lennard-Jones(LJ)12-6势,公式如下:
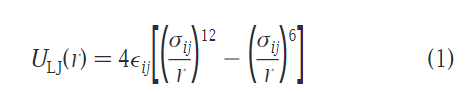
其中r为原子对距离,ε(字母代考)与σ为势能参数,因原子的种类而异,具体可以查看陈正隆老师的分子模拟一书。在martini力场中统一为σ=0.47nm,除了两个特别的类rings和antifreeze以外,还有一个例外就是Q类型和大多数极性类型 ,其C1和C2 斥力σ范围扩展为0.62nm。这个变化利于带电粒子在非极性介质中利于生成水化层。完整的参数数据如下表:

除了LJ势能以外,电荷组(类型Q)还有完整的qi,j联系,通过库能势:
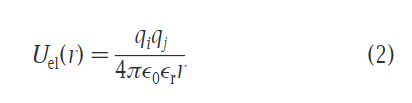
库能势与之前的比较准确性略有降低,主要是为了和之前的模型相比平衡增加的疏水强度。εr(字母代考)相对介电常数为15,与之前相比略微降低。
键联系:
键的描述和上一个版本相比没有升级,主要还是微弱的简谐振动提供的。此项在陈正隆老师的书籍版本中译为键伸缩势能项。平均键长与σ相同,为0.47nm,约束力为Kbond=1250Kj*mol-1,公式如下:
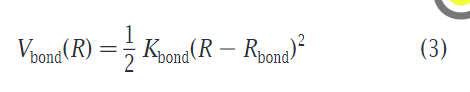
键角的简谐震荡项,参数依据为与全原子模型相互比较,计算公式如下:
[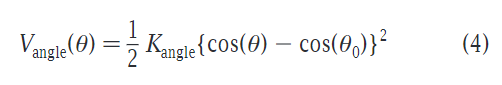
一般情况下Kangle=25KJmol-1,θ0=180°,在涉及cis double bond时,参数为Kangle=45KJmol-1(之前参数为35KJmol-1),θ0=120°,因为和全原子相比联系过于微弱。
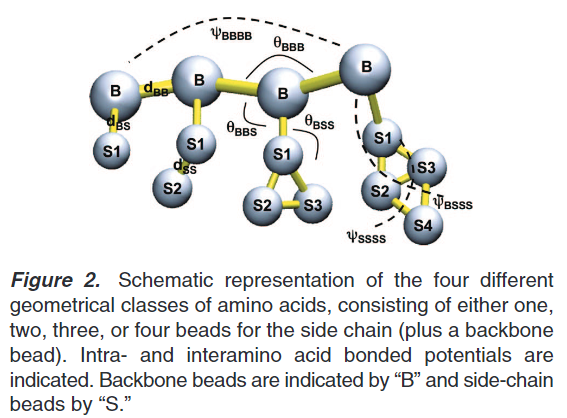
蛋白的键联系:
蛋白的键长,键角,二面角和他们各自的力场约束统计起来作为键参数的参考。这些参数来源于PDB库中近2000个蛋白作为数据集。蛋白的二级结构来源于DSSP项目。计算的键长,键角和二面角如下图:
PDB数据库中的参数分布如下图:
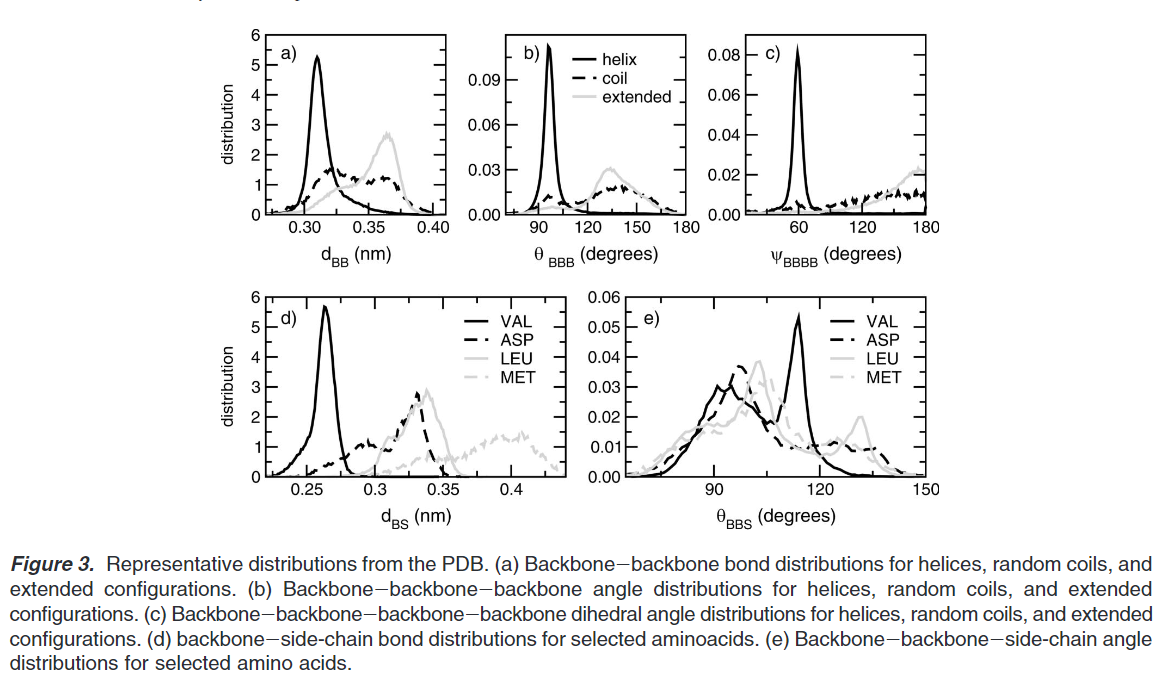
环化物参数:
对于环化物,4个原子称为1个珠子是不怎么适用的,最佳为2个或者3个位1个珠子,具体可以查看文章中给的环己烷,苯环等的例子。由于会照成很大CG珠子密度,因此这些参数需要差别对待。特别的设置标签为“S”,其LJ势能的σ也是不同的,σ=0.43nm,ε(字母代考)缩小至75%。环的话分子内的LJ不适合该体系,而更换improper dihedral angle势来合理的解释。其公式如下:
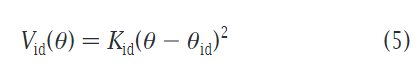
θ的角度由i,j,k和j,k,l构成,θid则为平均角度,力场约束为Kid。
二面角(dihedral angle)参数(蛋白):
对于氨基酸残基还有两个二面角势,其中improper 二面角势 Vid主要被用来阻止平面组的面外扭曲。Proper二面势Vd被使用来约束肽骨架的二级结构。这是一个非常重要的点,因此,若有蛋白序列二级结构的改变是不适合这个模型的(分别为公式5,6),公式如下:
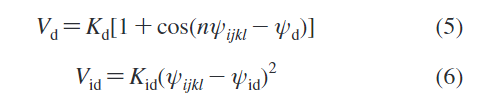
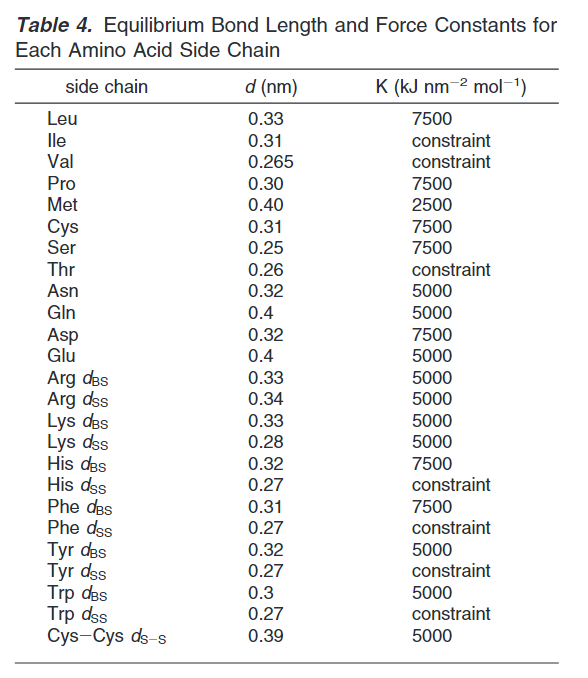
二面角参数仅仅在四个联系的珠子拥有相同的二级结构(螺旋或者延长(extended)),下表统计了所有侧链的键长和联系约束。
键角统计如下表:
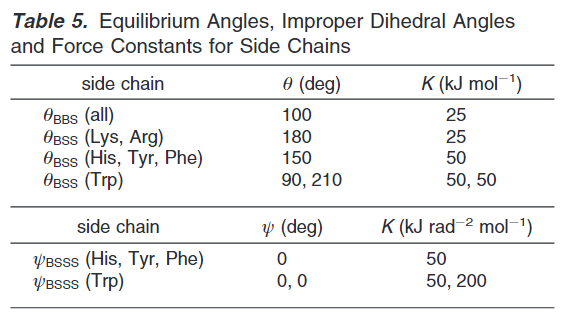
抗凝剂参数:
由于水模型是P4颗粒,相对于真实的水更容易freezing,故需要Antifreez参数。CG 水在280到300K可能会freezes。freezing是一个核驱动的过程,一般CG水能够保留其流动性(fluid)非常长的时间,但是一旦成核形成,将会很快速的形成,在固态表面甚至膜表面可能都会加速水的freeze。若研究更低温度同样如此,故需要增加水的无序程度。引入抗凝剂BigP4,LJ参数从0.47nm改为0.57nm,为了避免相的分离,BP4-P4联系上升为等级“O”。其他按照正常颗粒。当加入抗凝剂以后会对液体平衡性产生一定的影响,但作者对膜水体系进行分析,发现在膜表面积,形成gel phase的转移温度,膜分子的横向扩散长度都没有很大的影响,故认为为可接受范围之内。
模拟参数:
对于模拟参数,非键联系的cut off距离设置为rcut=1.2nm。为了避免产生不必要的噪音,使用GROMACS 的标准偏移功能,其中能量和约束力设置在截止距离处消失。LJ势能的偏移从rshift=0.9nm到rcut。静电势偏移从rshift=0.0nm到rcut。静电势的偏移会对模拟距离依赖性的screening造成影响。neighbor list可以升到10 steps, neighbor list cutoff设置等于rcut。模拟步长可以升至dt=40fs。当然设置成25fs到30fs可能会更加稳定。选择时间步长的不同对自由能有一定的影响,影响约为5%。(从40到5fs),所以说影响不是很大。
对于粗粒化模拟时间因子一般扩大为2到10倍,文献中默认的时间因子为4(注:文献1如是说,文献3说的为2到8倍,但时间因子均说的为4),即CG模型模拟10ns,实际上相当于现实中40ns,可以粗略的乘以4。产生这个现象的原因是CG模型相对于AA模型联系更加的平滑。
蛋白模拟:
文献中提供的作者使用的模拟参数如下,可能和官网的有出入,毕竟时间和版本较久(文献中的gromacs为3.3),故以下内容是过时的,仅供参考和笔记作用。
- 拓扑文件使用官网的脚本
- 每个组(lipids,water 和proteins)的温度算法采用Berendsen温度耦合算法,时间为1ps
- 使用Berendsen算法进行半均匀( Semi-isotropic)压力耦合,大气压为1bar任意的在膜平面上且垂直于膜,使用的时间约束为5.0ps,压缩率为4.5*10-5 bar-1
- 芳香氨基酸残基Val,Ile和Thr侧链和骨架侧链键使用的约束算法为LINCS算法,主要是为了避免快速波动(fluctuations,看到这个单词是否想到了RMSF?)所造成的大量的不稳定现象的发生。
- neighbor list 设置为10 steps,neighbor list cutoff等于rcut=1.2nm和其他设置为一样
- 由于计算影响的原因(猜测是为了更快),所有的珠子设置的相对质量为72,依据为4个水分子,对于环(ring)结构,相对质量设置为45
- 时间尺度和前面一样,时间因子为4,即25fs相当于有效时间100fs,当然这个估计是不精确的,故需要进行精确时间的模拟计算是行不通的,但是精确模拟计算运用是较少的,故对于研究扩散的时候要特别小心,即时有些扩散的CG模型研究与实验吻合较好。
拓扑中离子需要注意:
对于粒子,考虑到AA力场对ions难度已经很大,CG ion力场仅在定性上准确。
这段我也不是很懂,原文如下:
Keeping in mind the difficulty of model-ing of ions already with AA force fields, the CG ion force field is only qualitatively accurate
Martini力场性能测评部分结果:
氨基酸残基侧链在DOPC膜上跨膜研究
对不同侧链从DOPC膜中拉出(PMF)研究,将CG模型和全原子模型进行比较,研究结果表明疏水性氨基酸与全原子模拟相比有非常好的吻合,特别是barrier方面。如图中的leucine,isoleucine和valine。对于tryptophan和tyrosine,barrier在全原子profiles中不存在,且在CGprofiles低于5KJ/mol。且最小区域和全原子模拟拟合较好。对于极性氨基酸残基,对于全原子模型和CG模型,也有合理的分布。电荷残基在全原子模拟和CG模拟之间显示了非常大的不同。对于CG模型,其在进入膜中的自由能惩罚被严重低估,但是还是仍然高的(glu,asp和arg超过40,lys超过35)进入膜的数量是可以忽略不计的,换而言之其对于入膜这个时间是没有影响的,但是其他影响是有可能的,这也是力场需要改进的地方。完整的PMF结果如下图:
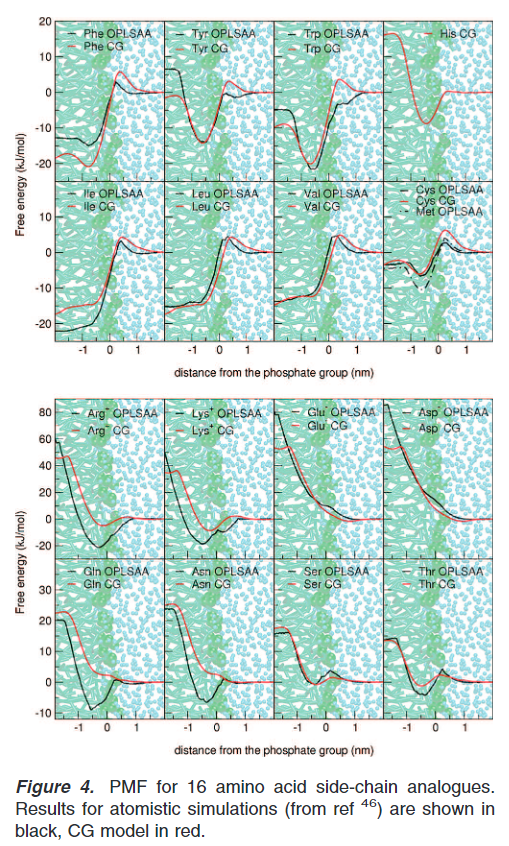
但是需要注意的是与其相比较的全原子力场为OPLSAA力场,OPLSAA在全原子力场中对于蛋白的表现也并不是那么的好,其对于蛋白是用的老的amber力场,且开发的人较少,长时间未开发(来源:GROMACS中文组-李继存老师)
氨基酸残基联系常数:
氨基酸残基i和j的定义如下:

其中1 / C是校正系统中物种浓度的因子,P(x)是在X状态下找到的概率。其中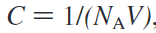

可以发现CG模型和全原子模拟的比例是一样的,但是CG模型较全原子模型数值有低估。
Martini力场的限制:
1.参数对于固体相并不是那么准确,相比液体相来说固体和气体相显得太稳定
2.内在熵变会丢失,温度依赖性受到了影响。Martini模型,也许在大多数CG方法中一样遇到的另一个困难是将极性和带电荷的化合物分配成低介电常数。因为极性物质的相互作用强度在非极性环境中会被低估。
3.由于开发的初衷是涉及在非极性环境中形成极性/带电复合物的应用尤其容易受到影响
4.忽略了长程电荷力是另一个限制,超过1.2nm的长程电荷不会被统计!但原则上是可以增加长程电荷的,必须认识到,静电相互作用方案的改进可能影响其他性质,例如每个脂质的面积或自发曲率
5.由于氨基酸残基二面角的存在,使得蛋白的二级结构无法改变,进行模拟的前提之一是蛋白二级结构是没有变化的,若需要查看二级结构的变化,可以使用Go模型。具体可以查看参考文献2.
由于这些限制,个人看法Martini力场还是有很多可以开发和提升的地方,这个力场还是很不是完美,特别是对于带电性质的研究,Martini力场可能是不可行的,有许多需要做的工作来进行完善,否则很难得到自己想要得到的结果!
最后更新于2017-9-30
参考文献:
- Marrink S J, Risselada H J, Yefimov S, et al. The MARTINI force field: coarse grained model for biomolecular simulations[J]. The journal of physical chemistry B, 2007, 111(27): 7812-7824.
- Go N, Taketomi H. Respective roles of short-and long-range interactions in protein folding[J]. Proceedings of the National Academy of Sciences, 1978, 75(2): 559-563.
- Monticelli L, Kandasamy S K, Periole X, et al. The MARTINI coarse-grained force field: extension to proteins[J]. Journal of chemical theory and computation, 2008, 4(5): 819-834.

