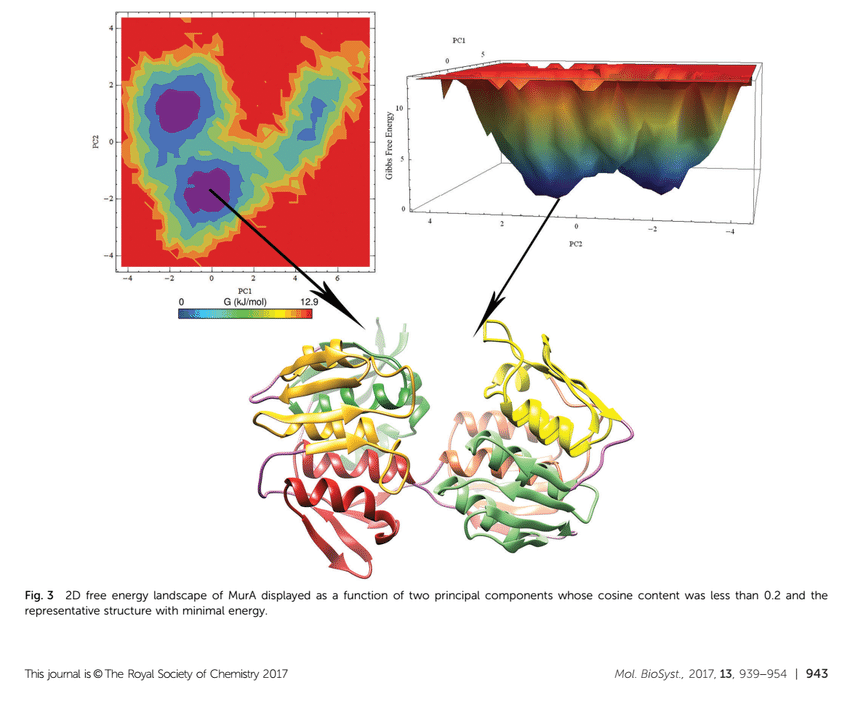
之前看到文献中有能量景观图,非常羡慕,而且感觉很高大上,等自己真正体会了主成分分析以后就比较释然了,但是若本身不是做模拟的,用来放一张图在支撑材料我感觉还是非常的好。
示例图如下:
图片1
比对结构
Tip:
之前一直卡在后面计算时间很长,后来才发现是未进行蛋白比对的缘故,若没有进行蛋白比对(即蛋白不乱动),请先蛋白比对一下,命令如下:
1
| gmx trjconv -f md.xtc -s md.tpr -fit rot+trans -o mdfit.xtc
|
生成协方差
首先我们需要使用gromacs covar来生成协方差矩阵并计算本征向量和本征值:
1
| gmx covar -s md.gro -f mdfit.xtc -o eigenvalues.xvg -v eigenvectors.trr -xpma covapic.xpm
|
运行后会询问选择哪些原子范围做比对,选择C-alpha,之后会询问选择哪些原子范围做PCA,选择C-alpha或者其他均可。
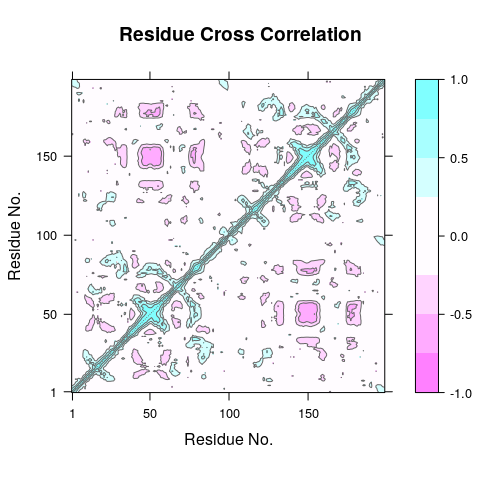
最后会生成协方差的图形。但是这个最好在bio3d中,效果更佳,如下图所示:
图片2
蓝色表示正相关,红色表示负相关,只需要看分界线的一面即可。
投影本征向量
若需要看PC1和PC2的投影,可以执行:
1
| gmx anaeig -f mdfit.xtc -s md.gro -v eigenvectors.trr -first 1 -last 2 -2d 2d.xvg
|
同样bio3d上面可能好看那么一丢丢:
但是这样的缺点就是会丢失时间系数,所以我们只能一个PC一个PC的倒出:
1 2 3
| gmx anaeig -f mdfit.xtc -s md.gro -v eigenvectors.trr -last 1 -proj pc1.xvg gmx anaeig -f mdfit.xtc -s md.gro -v eigenvectors.trr -frist 2 -last 2 -proj pc2.xvg
|
当然也可以写在一条里面。
再用perl脚本进行合并:
1
| perl sham.pl -i1 pc1.xvg -i2 pc2.xvg -data 1 -data2 1 -o gsham_input.xvg
|
再使用sham工具计算Gibbs自由能:
1
| gmx sham -f gsham_input.xvg -ls FES.xpm
|
生成的FES.xpm文件可以在linux系统下直接查看,但是不美观,我们可以使用脚本转换为文本文件用其他软件做图:
[注意该脚本为python2版本]
1
| python2.7 xpm2txt.py -f FES.xpm -o free-energy-landscape.txt
|
最后可以进行绘图,我是使用的matplotlib[注意我这里用的python3版本]:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22
| import pandas as pd import matplotlib.pyplot as plt import numpy as np data=pd.read_csv('free-energy-landscape.txt',sep='\t',header=None) fesvalue=np.array(data[2]) plotvalue=np.reshape(fesvalue,(32,-1)) plt.figure(figsize=(8,8)) plt.imshow(plotvalue,cmap='jet',interpolation='bilinear') plt.xticks([],[]) plt.yticks([],[]) plt.xlabel('PC1',fontsize=16) plt.ylabel('PC2',fontsize=16) plt.savefig('fes.png',dpi=600)
|
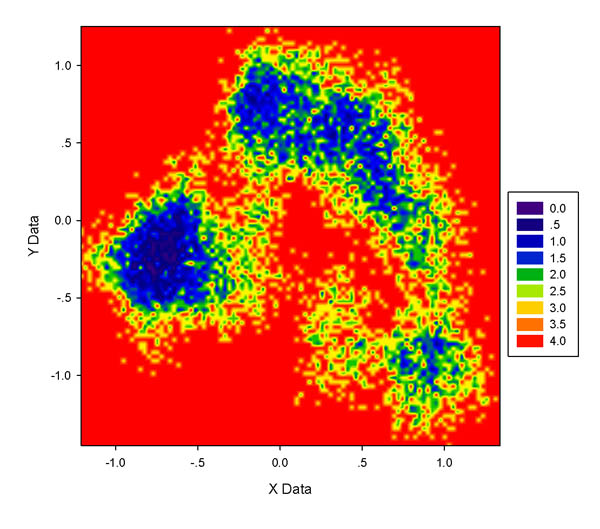
由于文章未发表,所以盗取sob老师的一张图,大致也差不太多:
图片3
参考资料:
Free Energy Landscape- PCA
浅谈PCA与g_covar+g_anaeig+ddtpd+sigmaplot做自由能面图的方法
GROMACS分子动力学模拟教程:多肽-蛋白相互作用